Hit Discovery and Structural Design Group
Dr Rob van Montfort’s group uses screening techniques to narrow down the number of potential molecules to take forward into drug development.
Research, projects and publications in this group
Our group establishes, validates and runs high-throughput and fragment screens on cancer drug targets while also characterising protein-ligand interactions of hit matter resulting from our fragment and high-throughput screens.
This group consists of the Analytical Technology and Screening and the Structure-Based Drug Design research groups:
One of the key steps in target-directed cancer drug discovery is the identification of small molecule compounds (hits) that inhibit the activity of validated target proteins. This can be achieved by high-throughput screening of large lead-like compound collections using appropriate biochemical and cell-based assay formats that report the activity of the drug target, or by screening small compound libraries of low molecular weight molecules (fragments) using very sensitive biophysical techniques and high concentration biochemical assays.
The role of the Analytical Technology and Screening group is to establish, validate and run high-throughput and fragment screens on cancer drug targets under study at the Centre for Cancer Drug Discovery, and to carry out the initial characterisation of the hits obtained. Presently, we have a screening library of approximately 220,000 lead-like compounds, which we developed with our partner organisation Cancer Research Technology.
In addition, we have developed a fragment library of more than 2,000 compounds. Once progressible compound series have been identified, the group is involved in informing medicinal chemistry aimed at understanding and improving the pharmaceutical properties of the hits generated.
In addition, mechanistic endpoint assays are developed to support the CTU's drug discovery projects into clinical evaluation. These assays include, high-throughput phenotypic assays based on standard ELISA methodology and high content imaging assays based on the use of the InCell analyser.
The Structure-Based Drug Design Group is a joint initiative between the Divisions of Structural Biology and Cancer Therapeutics and consists of protein biochemists and protein crystallographers. The group has strong links with Medicinal Chemistry, In Silico Medicinal Chemistry and Biology teams within the CTU, and with the other crystallography team within the Division of Structural Biology.
The group uses biophysical techniques such as Tm-shift assays, Surface Plasmon Resonance, Isothermal Titration Calorimetry and High-Throughput X-ray Crystallography to characterise protein-ligand interactions of hit matter resulting from our fragment and high-throughput screens and continue to investigate these interactions as the hits progress to advanced inhibitors.
The activities of the Hit Discovery and Structural Design Team are underpinned by a state-of-the art protein production capability which is responsible for cloning, expression and purification of recombinant protein targets for the biophysical characterisation and X-ray crystallography as well as for the biochemical assays carried out in the Analytical Technology and Screening group. The team is experienced in construct design and has its own E. coli and insect cell expression facilities and a protein purification laboratory.
Research projects
Inhibitors of monopolar spindle 1 (MPS1) kinase
The protein kinase MPS1 is a crucial component of the spindle assembly checkpoint signal and is aberrantly overexpressed in many human cancers. MPS1 is one of the top 25 genes overexpressed in tumors with chromosomal instability and aneuploidy; PTEN-deficient breast tumor cells are particularly dependent upon MPS1 for their survival making it a target of significant interest in oncology.
We are collaborating with Professors Spiros Linardopoulos, Julian Blagg and Dr Swen Hoelder and the CRT Pioneer fund LP to discover small molecule inhibitors of MPS1. Using a high throughput screen of an in-house kinase-focused library combined with a structure-based drug discovery approach, we have discovered CCT251455, a potent, selective and orally bioavailable chemical tool for MPS1. Interestingly, CCT251455 stabilizes an inactive conformation of MPS1 with the activation loop ordered in a manner incompatible with ATP- and substrate peptide–binding (Fig 1).
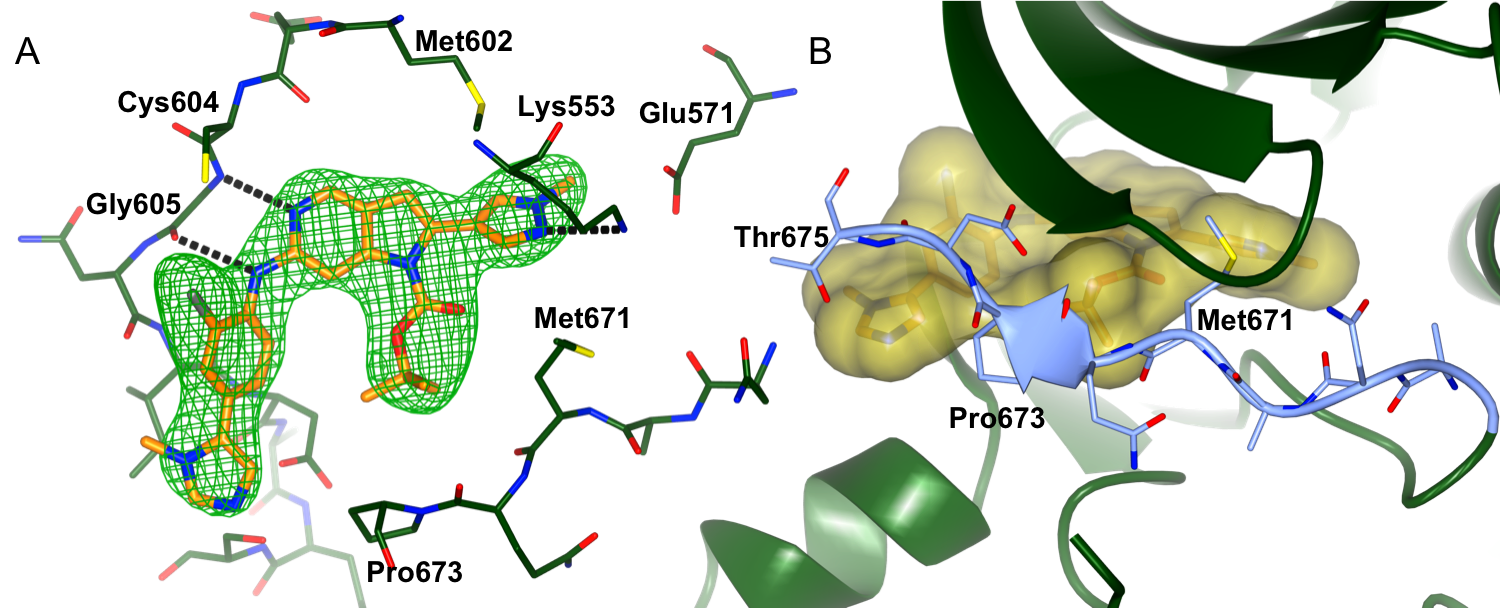 Figure 1: Binding of CCT251455 to MPS1. (A) Binding of CCT251455 to the hinge region of MPS1. (B) CCT251455 stabilises and unusual conformation of the activation loop (light blue).
Figure 1: Binding of CCT251455 to MPS1. (A) Binding of CCT251455 to the hinge region of MPS1. (B) CCT251455 stabilises and unusual conformation of the activation loop (light blue).
Inhibitors of the KDM4/5 JmJC histone demethylases
Histone demethylases are involved in the regulation of the methylation status of lysine residues on the N-termini of DNA-bound histones, a process increasingly implicated in cancer. The JmjC histone demethylase proteins are a family of Fe(II) and 2-oxoglutarate (2-OG)-dependent enzymes which convert trimethylated lysine substrates to their dimethylated forms. The KDM4 JmJC demethylase subfamily consists of six members.
In collaboration with Professor Julian Blagg and the Structural Genomics Consortium (SGC, Oxford) we aim to discover novel and inhibitors selective for the KDM4 subfamily. From a high-throughput screen followed by a structure-based drug discovery approach we have discovered potent inhibitors selective for the KDM4 and KDM5 histone lysine demthylases (Fig. 2).
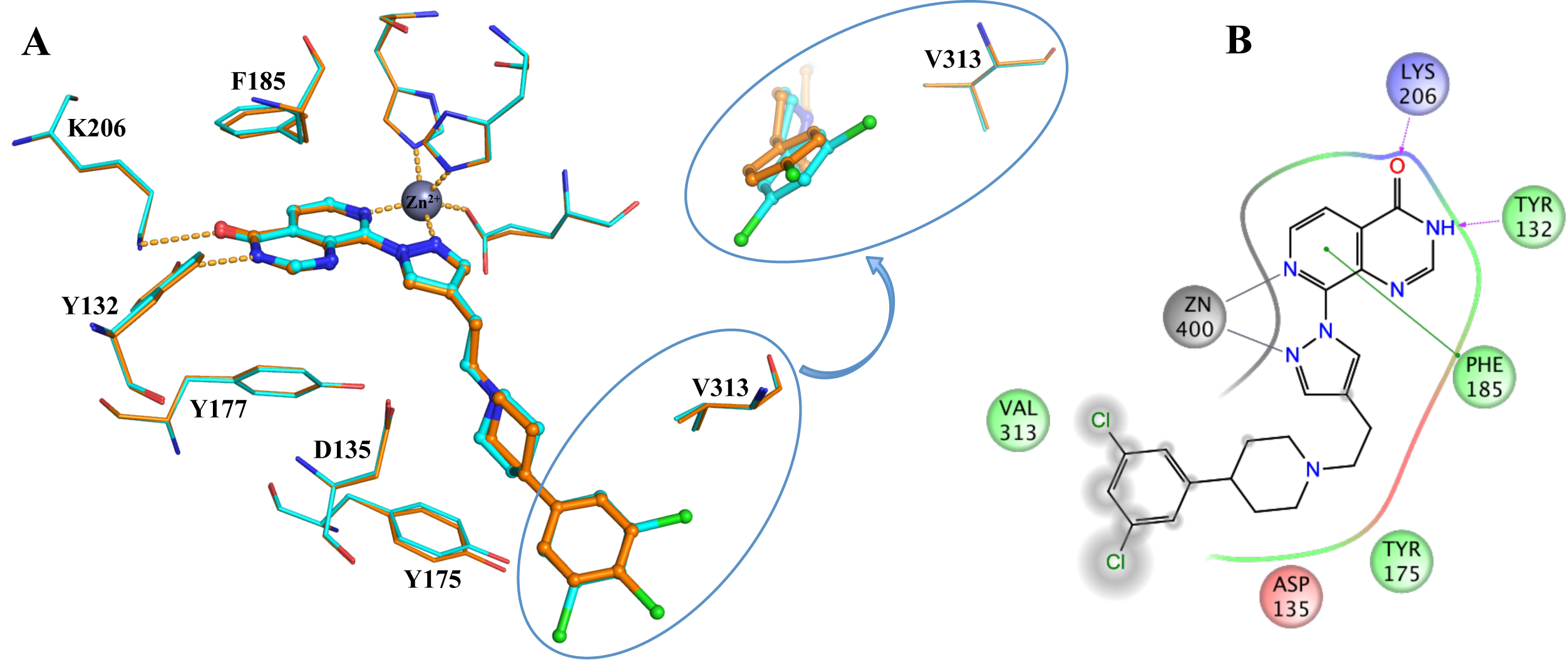 Figure 2: Binding of KDM4/5 inhibitors to KDM4A. A) Superposition of two KDM4/5 inhibitors (blue/organge) bound to KDM4A highlighting key interactions with the protein. B) 2D interaction diagram showing the interactions of the most potent inhibitor (orange) with KDM4A.
Figure 2: Binding of KDM4/5 inhibitors to KDM4A. A) Superposition of two KDM4/5 inhibitors (blue/organge) bound to KDM4A highlighting key interactions with the protein. B) 2D interaction diagram showing the interactions of the most potent inhibitor (orange) with KDM4A.
AGC kinase inhibitors
Diazaspirocyle-based PKA inhibitors
One of the best protein kinase in the AGC kinase family is the cAMP-dependent protein kinase A (PKA). As part of a collaboration with Professor Ian Collins to evaluate substituted diazaspirocycles as scaffolds to probe the ATP-binding site of protein kinases, we determined high-resolution protein-ligand structures of several diazacpirocycle-based inhibitors (Fig 1).
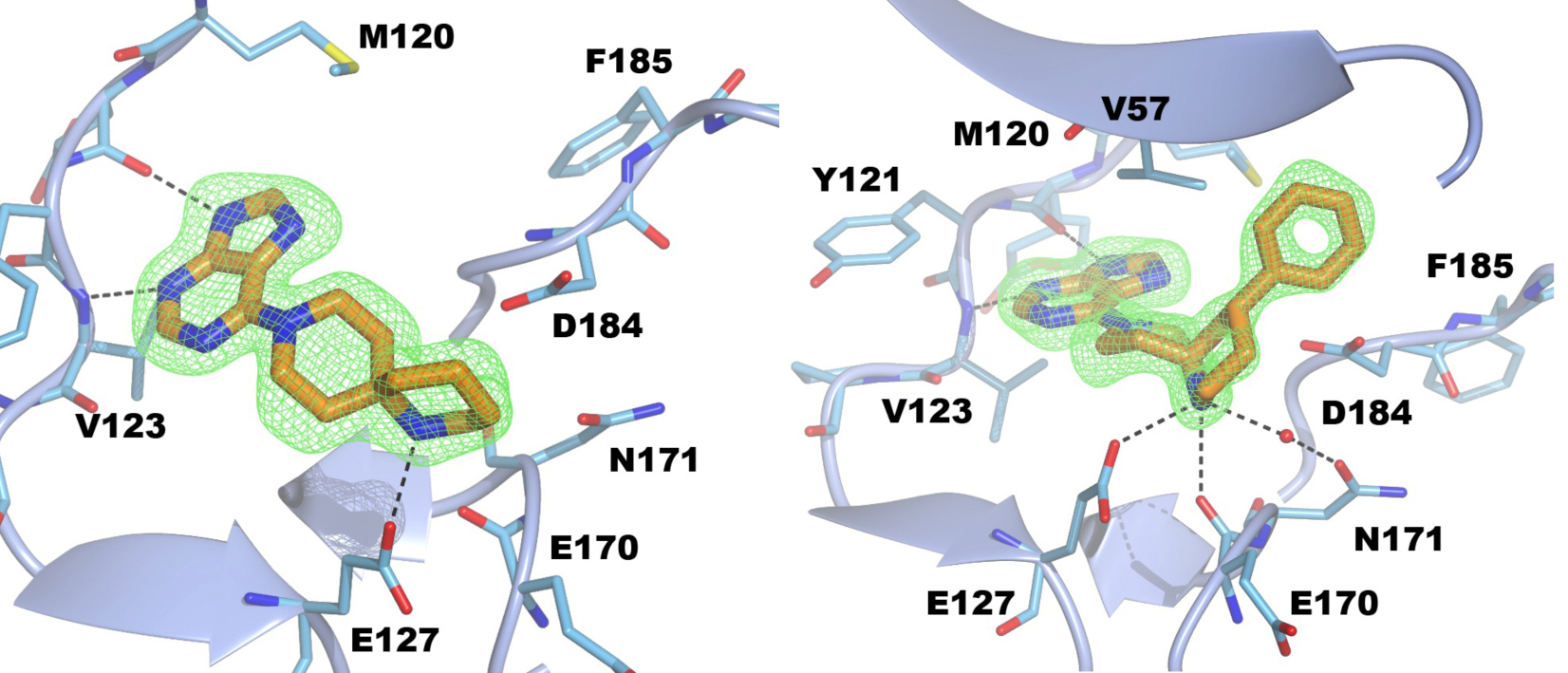
Figure 3: Crystal structures of PKA in complex with two different spirocycle-based inhibitors.
S6K inhibitors
The 70 KDa ribosomal S6 kinases (S6K) RPS6KB1 (S6K1) and RPS6KB2 (S6K2) are key effectors of PI3K/mTOR-regulated signalling, and have been implicated in a variety of human diseases including diabetes and cancer. They belong to the family of AGC kinases and are highly homologous with a sequence identity of 83% in their catalytic domains. We are not pursuing the discovery of S6K inhibitors for the treatment of cancer, but our crystallography efforts provide an example of the use of chimeric proteins as structural surrogate systems in structure-based drug discovery. Currently, no apo-structures of S6 kinases are avaliable due to a susceptibility of the apo-enzyme to aggregation. Reported S6K1 structures are protein-ligand complexes with the pan-kinase inhibitor staurosporine, making the system unsuitable for iterative protein-ligand crystallography to study ATP binding site inhibitors.
To generate a robust crystal system suitable for the generation of high-resolution protein-ligand structures to facilitate S6K inhibitor design, we developed a S6K1 chimeric protein based on the catalytic subunit of the closely related AGC kinase PKAα (PKA, Fig. 4).
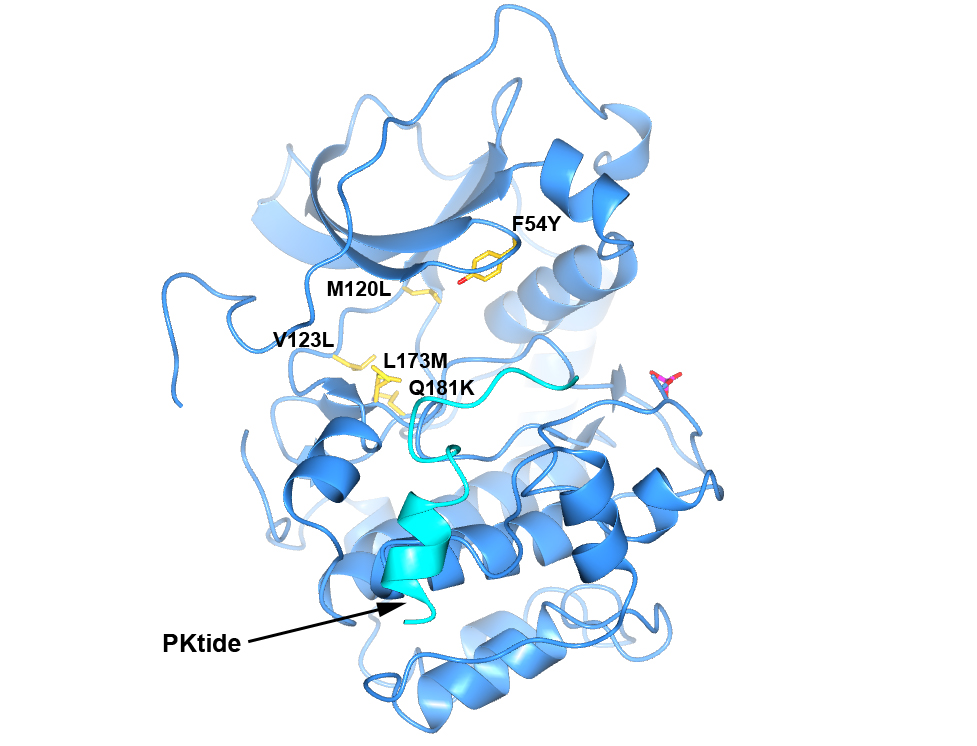 Figure 4. Structure of PKA with the five mutations in and near the ATP-binding site highlighted in yellow. The PKtide substrate inhibitor is shown in light blue
Figure 4. Structure of PKA with the five mutations in and near the ATP-binding site highlighted in yellow. The PKtide substrate inhibitor is shown in light blue
S6K1 and PKA share a sequence identity of approximately 33% in their kinase domains and their ATP-binding sites are nearly identical. Our PKA-S6K1 chimera was created by mutation of five residues in or near the ATP-binding site (F54Y, M120L, V123L, L173M and Q181K) that differ between the two kinases. Using this chimera we could obtain high-resolution structures of two inhibitor series identified in our high-throughput screen carried out in collaboration with Professor Keith Jones and scientists from Newcastle University (Fig. 5).
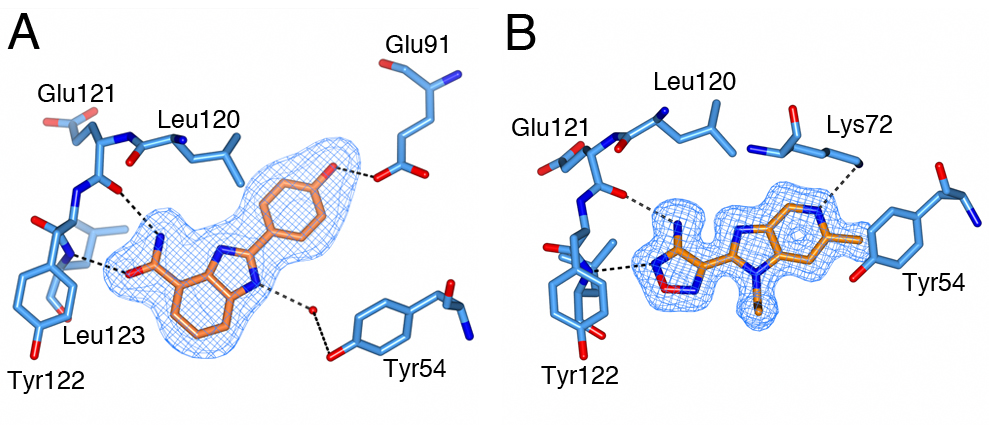 Figure 5. S6K inhibitors bound in the PKA-S6K1 chimeara. A) High resolution structure (2.2Å) of a carboxamidobenzimidazole HTS hit bound in the PKA-S6K1 ATP binding site. B) High resolution structure (1.6Å) of a benzimidazole oxadiazole S6K inhibitor bound in the PKA-S6K1 ATP binding site.
Figure 5. S6K inhibitors bound in the PKA-S6K1 chimeara. A) High resolution structure (2.2Å) of a carboxamidobenzimidazole HTS hit bound in the PKA-S6K1 ATP binding site. B) High resolution structure (1.6Å) of a benzimidazole oxadiazole S6K inhibitor bound in the PKA-S6K1 ATP binding site.
Fragment screening of Checkpoint kinase 2
Checkpoint kinase 2 (CHK2) is a serine/threonine kinase which is crucial in the activation of signal transduction pathways involved in the cellular response to DNA damage caused by external agents. The therapeutic value of CHK2 inhibition is still unclear, but selective CHK2 inhibitors could be potentially beneficial in a variety of contexts.
Professor Michelle Garett (University of Kent, previously at The Institute of Cancer Research) has shown that, while CHK2 inhibition did not potentiate the effect of DNA-damaging chemotherapy, it did sensitize cancer cells to the effects of PARP inhibitors that compromise DNA repair.
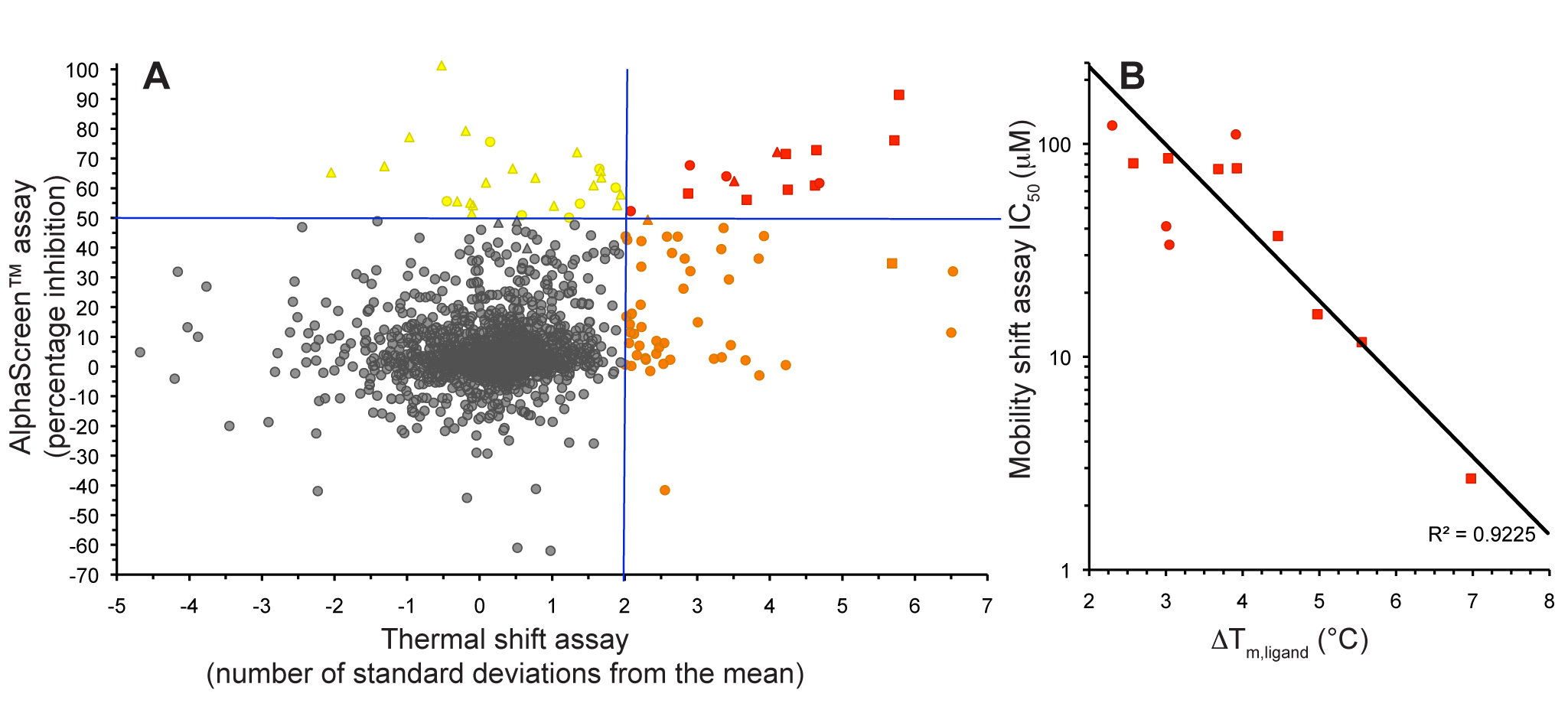 Figure 1. Fragment screening data from biochemical and thermal shift assays. (A) Comparison showing the primary AlphaScreen™ data plotted along the vertical axis as percentage inhibition, and the thermal shift data plotted along the horizontal axis as the number of standard deviations from the mean Tm, ligand for each plate and thresholds displayed as blue lines Hits in AlphaScreen™ and thermal shift are displayed in yellow and orange respectively. Mutual hits are shown in red, inactive fragments are grey. (B) Comparison of the IC50 and DTm, ligand values for the screening hits. Fragments showing interference in the AlphaScreen™ are indicated as triangles. Fragments confirmed in crystallography are shown as squares.
Figure 1. Fragment screening data from biochemical and thermal shift assays. (A) Comparison showing the primary AlphaScreen™ data plotted along the vertical axis as percentage inhibition, and the thermal shift data plotted along the horizontal axis as the number of standard deviations from the mean Tm, ligand for each plate and thresholds displayed as blue lines Hits in AlphaScreen™ and thermal shift are displayed in yellow and orange respectively. Mutual hits are shown in red, inactive fragments are grey. (B) Comparison of the IC50 and DTm, ligand values for the screening hits. Fragments showing interference in the AlphaScreen™ are indicated as triangles. Fragments confirmed in crystallography are shown as squares.
In collaboration with Professors Ian Collins and Michelle Garett we conducted a fragment screen against CHK2 using a combination of a high-concentration Amplified Luminescent Proximity Homogeneous Assay Screen (AlphaScreen™) kinase assay and a thermal shift assay. We showed that the use of these orthogonal techniques allowed efficient discrimination between genuine hit matter and false positives from each individual assay technology (Fig. 1).
We identified a number of chemically different ligand-efficient CHK2 hinge-binding scaffolds that have not been exploited in known CHK2 inhibitors. In addition, the CHK2 crystal structures with a quinoxaline-based fragment and its follow-up compound highlight a hydrophobic area above the hinge region not previously explored in rational CHK2 inhibitor design, but which might be exploited to enhance both potency and selectivity of CHK2 inhibitors.
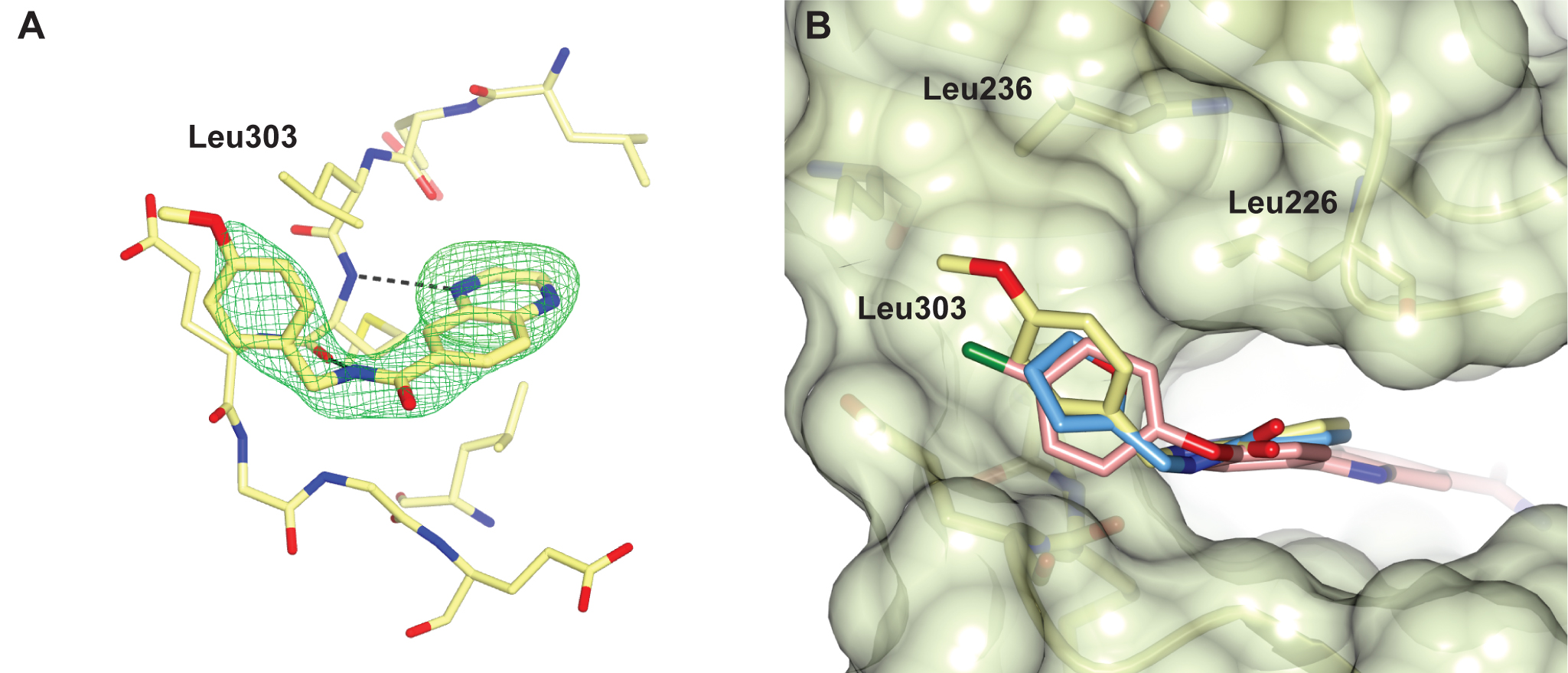
Figure 2 A selectivity pocket above the hinge. (A) Quinoxaline fragment follow-up compound binding to the CHK2 hinge region. (B) Superposition of the quinoxaline fragment (light-blue), its follow-up compound (yellow) and the arylbenzimidazole CHK2 inhibitor (pink, PDB ID code 4A9R), showing the fragments bind in a nearly identical manner with their respective furan and p-methoxyphenyl group binding in a hydrophobic pocket above the hinge, which is also accessed by the chlorophenyl group of the benzimidazole-based CHK2 inhibitor.
A fragment-based approach to target the molecular chaperone HSP70
The 70 kDa heat shock proteins (HSP70s) are an abundant family of ATP-dependent molecular chaperones that are crucial in cellular processes including protein folding, prevention of protein aggregation, modulation of protein complexes, and protein transport between cellular compartments. They have been implicated in several diseases, including cancer and are of significant interest as targets for novel cancer therapies.
In collaboration with Professors Paul Workman and Ian Collins we are applying fragment and structure-based approaches (Fig 3A). HSP70 are extremely flexible proteins (Fig. 3B), therefore a key challenge is to understand how the conformational flexibility of HSP70s affects ligand binding and how it can be exploited in inhibitor design.
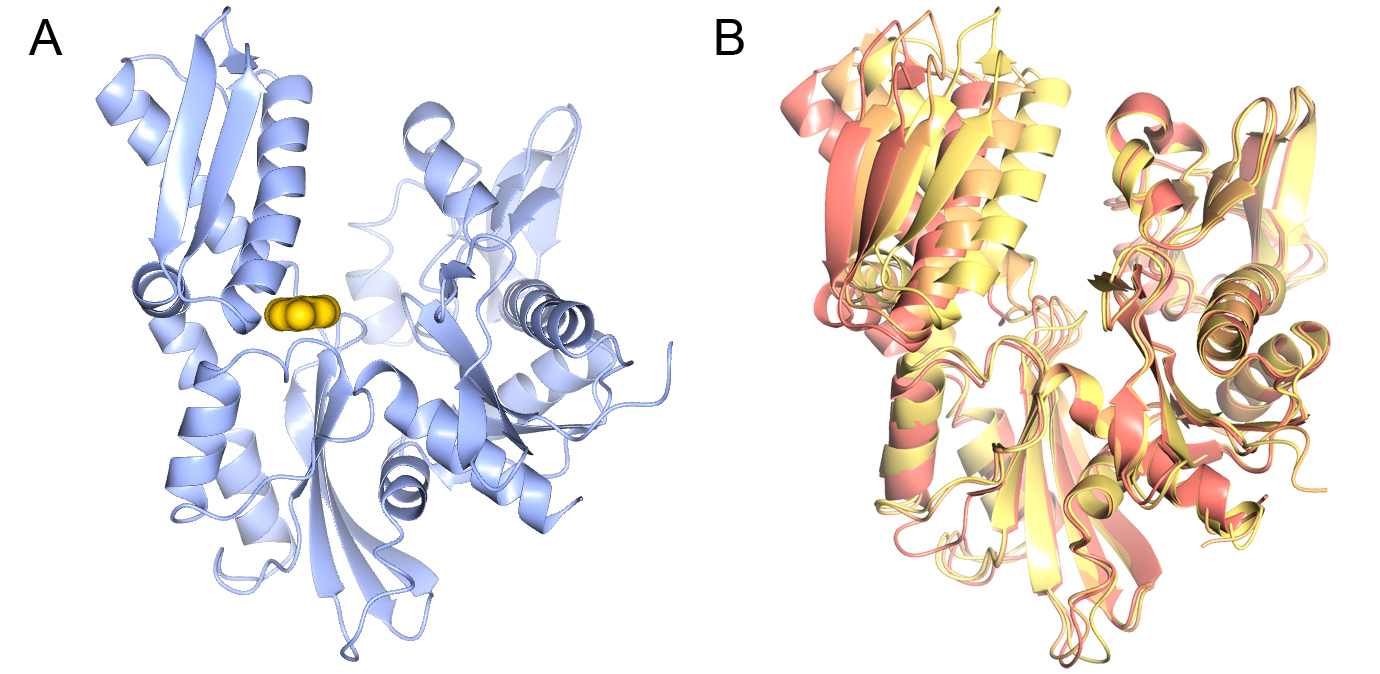
Figure 3 Structure of the HSP70 nucleotide-binding domain. A) A fragment bound in the ATP binding site of the HSP70 nucleotide-binding domain. B) Conformational flexibility on the HSP70 nucleotide-binding domain.
Identification of IRE1 inhibitors using a 384-well DELFIA assay
Inositol-requiring enzyme 1 alpha (IRE1α) is a transmembrane sensor protein with both kinase and ribonuclease activity which plays a crucial role in the unfolded protein response (UPR).
Protein misfolding in the endoplasmic reticulum (ER) lumen triggers dimerisation and subsequent trans-autophosphorylation of IRE1α. This leads to the activation of its endoribonuclease (RNase) domain and splicing of the mRNA of the transcriptional activator XBP1, ultimately generating an active XBP1 (XBP1s) implicated in multiple myeloma survival.
In collaboration with Professor Ian Collins and Dr Faith Davies and led by Dr Rosemary Burke, we have developed a 384-well screening assay using DELFIA® technology that is specific for IRE1α autophosphorylation. Using this format a focused library of 2312 potential kinase inhibitors was screened and several novel IRE1α kinase inhibitor scaffolds were identified (see Fig 1 below).
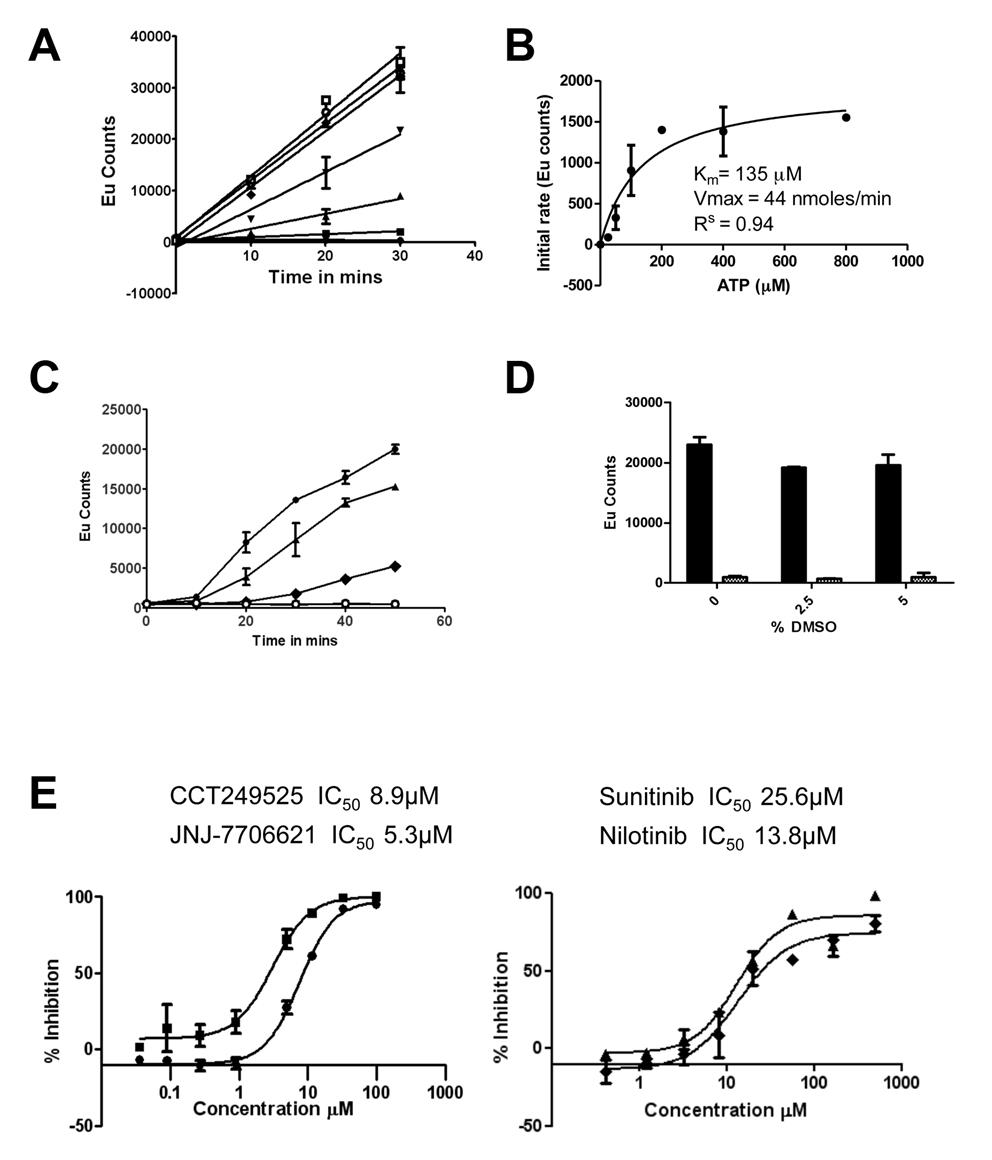 Figure 1. The IRE1α autophosphorylation DELFIA® assay. (A) Time course with varying ATP concentrations used to calculate the Km for adenosine triphosphate (ATP) (● no ATP, ■ 25 µM ATP, ▲ 50 µM ATP, ▼100 µM ATP,♦ 200 µM ATP, ○ 400 µM ATP,□ 800 µM ATP). B) Determination of the Km for adenosine triphosphate (ATP). C) Enzyme reaction time course; mean +/- SD of n=2 wells (● enzyme reaction at 700 nM IRE1α and 100 µM ATP, ▲ enzyme reaction at 500 nM IRE1α and 100 µM ATP, ♦ enzyme reaction at 350 nM IRE1α and 100 µM ATP,○). Enzyme reaction at 700 nM IRE1α without ATP). D) Effect of DMSO on the IRE1α autophosphorylation DELFIA®, showing no effect on the assay with up to 5% DMSO. E) Inhibition of IRE1α autophosphorylation activity by CCT249525 (●) , JNJ-7706612 (■), Sunitinib (♦) and Nilotinib (▲).
Figure 1. The IRE1α autophosphorylation DELFIA® assay. (A) Time course with varying ATP concentrations used to calculate the Km for adenosine triphosphate (ATP) (● no ATP, ■ 25 µM ATP, ▲ 50 µM ATP, ▼100 µM ATP,♦ 200 µM ATP, ○ 400 µM ATP,□ 800 µM ATP). B) Determination of the Km for adenosine triphosphate (ATP). C) Enzyme reaction time course; mean +/- SD of n=2 wells (● enzyme reaction at 700 nM IRE1α and 100 µM ATP, ▲ enzyme reaction at 500 nM IRE1α and 100 µM ATP, ♦ enzyme reaction at 350 nM IRE1α and 100 µM ATP,○). Enzyme reaction at 700 nM IRE1α without ATP). D) Effect of DMSO on the IRE1α autophosphorylation DELFIA®, showing no effect on the assay with up to 5% DMSO. E) Inhibition of IRE1α autophosphorylation activity by CCT249525 (●) , JNJ-7706612 (■), Sunitinib (♦) and Nilotinib (▲).
Dr Rob Van Montfort
Group Leader:
Hit Discovery and Structural Design
Dr Rob van Montfort uses protein crystallography and a range of biochemical and biophysical approaches to investigate ligand-binding to new cancer drug targets. He has a joint appointment between the Divisions of Cancer Therapeutics and Structural Biology at the Centre for Cancer Drug Discovery.
Researchers in this group
 .
.
 .
.
Email: [email protected]
Location: Sutton
Dr Rosemary Burke is a Cancer Research UK funded Senior Staff Scientist in the Hit Discovery and Structural Design Group at The Institute of Cancer Research, London with an important role managing the assay development and screening capabilities for the drug discovery projects in the Centre for Cancer Drug Discovery.
Dr Rob Van Montfort's group have written 135 publications
Most recent new publication 12/2024
See all their publicationsRecent discoveries from this group

