
At the 2015 NCRI Conference Professor Mel Greaves, our founding author, was given the Lifetime Achievement Award by CRUK
This post is a synopsis of the lecture I gave at the National Cancer Research Institute (NCRI) conference in Liverpool on the 2nd November 2015 – minus some anecdotes about mentors and colleagues – you needed to be there to hear those. It’s my personal, historical narrative of tackling the challenge involved in unravelling the biology and causes of childhood leukaemia.
Childhood acute lymphoblastic leukaemia (ALL) is the most frequent subtype of paediatric cancer in the developed world. Before the availability of effective treatment, it was universally fatal. The cure rate, from combination chemotherapy, now sits at around 90%.
But what is its cause and is there potential for prevention? The epidemiological literature on this topic provides a cornucopia of purported risk factors. Some are plausible, others fanciful flying pigs. Studying the possible causal exposures via case/control studies of a relatively rare (1:2000 risk for children) cancer that is biologically heterogeneous is tough, so perhaps we shouldn’t be too surprised at the paucity of robust data.
My take on this has been to value only epidemiological analysis that is contingent upon, or driven by, the underlying biology and to assume, as was argued 40 years ago by the epidemiologists Richard Doll and Richard Peto, that this cancer, like most, if not all, others will have a multi-factorial causal mechanism involving exposures, inherited susceptibility and chance. And, critically, I assume that the problem is likely to be informed by an evolutionary perspective.
My involvement with this disease began in the 1970’s when we established, using immunological markers and the then new technology of flow cytometry, that ALL has biologically distinct subtypes and that these were clinically different and possibly aetiologically distinct. This was a game changer. We now know that these subtypes reflect developmental, lineage origins, and are underpinned by distinct genetic abnormalities.
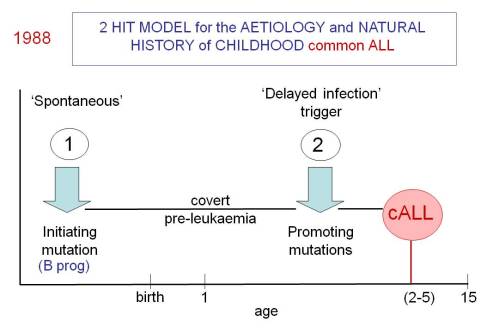
A striking feature of the most prevalent subtype that we dubbed ‘common ALL’ was its sharp age incidence peak between 3 and 5 years of age.
Through working with an international consortium of colleagues, we found that this age peak of diagnosis was seen in all developed societies but was diminished or absent in less affluent populations, e.g. black South Africans. Other subtypes of ALL, acute myeloblastic leukaemia (AML) and lymphoma in children did not present in this fashion.
The idea that leukaemia in children might be driven in some way by infection has been on the table for almost 100 years. But how odd that the most common type of ALL seems to have appeared at a time when the more serious infections of infancy and children had become greatly diminished?
So: why not turn the problem on its head? Maybe a lack of infection in infancy could be a factor? It’s not as bizarre as you might first suppose. The immune system has evolved as a network and in a way that it is non-hardwired at birth so is in need of modulation. This occurs, or used to occur, via common infections early in life, the impact of which is modulated by mothers’ immune status and breast milk.
A deficit of infectious exposure in infancy would, as animal studies indicate, leave the unprimed immune system at risk of very dysregulated responses to later infections – say when children start school and begin to be exposed via social mixing.
So here, in a nutshell, is the ‘delayed infection’ hypothesis for childhood ALL first proposed in 1988 on the back of an envelope (Slide 1). It’s not a million miles away from the ‘hygiene’ hypothesis that offers an explanation of why certain auto-immune diseases (type 1 diabetes and multiple sclerosis) and allergies are very prevalent in modern and ‘sanitised’ societies.
Slide 1
Validating this hypothesis has not been easy. It’s been a 25 year journey and the evidence is still incomplete. But I think it’s more or less correct.
The critical tests were:
- Establishing the ‘pre-‘ plus ‘post-‘ natal two hit hypothesis for common, pre-B cell precursor ALL. This required identifying the developmental timing of the common or recurrent mutations in ALL.
- Documenting that pre-natal initiation gives rise to a covert pre-malignant clone that will only convert to overt, clinical ALL in the context of additional genetic abnormalities.
- Determining the underlying mechanism that gives rise to the critical secondary mutations.
- Forging a link between common infections, the immune response and the biology of pre-leukaemic clones, i.e. their expansion and mutagenesis.
- Identifying the inherited alleles that modify risk of this clonal process occurring.
- Deriving evidence from appropriately designed epidemiological studies and animal modelling that supports the notion that common infections provide the essential ‘promotional’ drive for the key secondary (post-natal) infections in ALL.
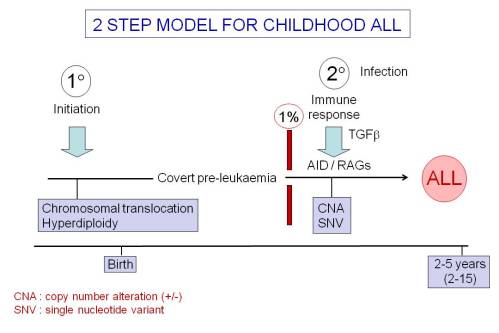
Establishing the historical order of genetic events that are present in a diagnostic sample was an interesting challenge. Tracking back the clonal, evolutionary past was achieved by using three different tactics (Slide 2). Collectively these confirmed that ALL usually originates by gene fusion (ETV6-RUNX1) or hyperdiploidy in utero but other recurrent gene mutations that are necessary for ALL to develop, principally copy number deletions, occur post-natally.

Slide 2
The comparative genomics of leukaemia both in twin pairs and single cell genetic analysis firmly place ETV6-RUNX1 fusion as the founder event at the root of the phylogenetic or evolutionary tree of clones or subclones in ALL.
The rest of the jigsaw has also fallen into place with respect to the underlying genetics and biology. Five or six relatively common allelic variants have been found, in genome wide association studies, to confer an inherited increase in risk (odds ratios of 1.2 – 1.5).
The recurrent copy number changes in ALL are driven by ‘off target’ mutagenesis by the recombinases RAG1/2 that, physiologically, rearrange IGH and TCR loci. But this requires the additional or concomitant expression of the cytosine deaminase enzyme AID.
Murine models confirm that ETV6-RUNX1 expression in the B cell lineage can give rise to B cell precursor ALL if, and only if, AID expression is activated by a surrogate signal of infection (bacterial LPS was used in the experiments, but IL4 and TGFβ will also do the trick). But there is no ALL in the murine model if either RAGs or AID are deleted from the genetic background.
TGFβ may be a key player and ‘missing link’ in this story. It selectively expands the clone size of murine and human pre-leukaemic cells expressing ETV6-RUNX1 and activates AID in human B precursor cells.
So, what’s still lacking? It’s back to epidemiology. If, as we suspect, it involves a variety of infections rather than a specific leukaemia virus (as is the case for leukaemia in cattle and cats), then providing unambiguous evidence is challenging.
One clear prediction of the ‘delayed infection’ hypothesis, however, is that infants with more social contacts in the first year of life should have a reduced risk of ALL. We found this to be the case in a large UK-based case/control study and studies in other countries (Scandinavia, USA and France). Meta-analysis combining this data confirms the significance of the observation.
What more might consolidate the story? Perhaps a more high fidelity model using xeno-transplanted human progenitor cells expressing ETV6-RUNX1 and AID. Occasional space/time clusters of childhood ALL do occur and in these, albeit rare, instances it might be possible to implicate a specific, though not transforming, microbial infection as a promoter of disease.
So: here’s a schematic of the current picture we have of the causal mechanisms, natural history and clonal evolution of ALL (Slide 3).
Slide 3
Mel
Key Papers
1. Greaves MF, Wiemels J (2003) Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer, 3: 639-649.
2. Greaves MF, Maia AT, Wiemels JL, Ford AM (2003) Leukemia in twins: lessons in natural history. Blood, 102: 2321-2333.
3. Greaves M (2006) Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer, 6: 193-203.
4. Bateman CM, Colman SM, Chaplin T, Young BD, Eden TO, Bhakta M, Gratias EJ, van Wering ER, Cazzaniga G, Harrison CJ, Hain R, Ancliff P, Ford AM, Kearney L, Greaves M (2010) Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood, 115: 3553-3558.
5. Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, Kinsey SE, Lightfoot T, Roman E, Irving JAE, Allan JM, Tomlinson I, Taylor M, Greaves M, Houlston RS (2009) Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nature Genet, 41: 1006-1010.
6. Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, Kearney L, Enver T, Greaves M (2011) Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature, 469: 356-361.
7. Papaemmanuil E, Rapado Martinez I, Li Y, Potter NE, Wedge DC, Tubio J, Alexandrov LB, Van Loo P, Cooke SL, Marshall J, Martincorena I, Hinton J, Gundem G, van Delft FW, Nik-Zainal S, Jones DR, Ramakrishna M, Titley I, Stebbings L, Leroy C, Menzies A, Gamble J, Robinson B, Mudie L, Raine K, O’Meara S, Teague JW, Butler AP, Cazzaniga G, Biondi A, Zuna J, Kempski H, Müschen M, Ford AM, Stratton MR, Greaves M, Campbell PJ (2014) RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet, 46: 116-125.
8. Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon S-M, Trageser D, Hasselfeld B, Henke N, Mooster J, Geng H, Schwarz K, Kogan SC, Casellas R, Schatz DG, Lieber MR, Greaves MF, Müschen M (2015) Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol, 16: 766-774.
comments powered by