
Cancers are life threatening because they migrate within the body, spreading far from their point of origin. This process – metastasis – hijacks tissues and compromises their critical functions. When they reach this stage, most cancer clones will be robust and resistant to treatment, whether that be radiotherapy, chemotherapy or immunotherapy. So, in a sense, it is resistance that is the major stumbling block to successful treatment. Those exceptional cancers that are curable, even when disseminated (childhood acute lymphoblastic leukaemia, testicular cancer and choriocarcinoma) retain sensitivity.
How do cancer cells acquire and express resistance and why is it so prevalent? It turns out that there are several distinct mechanisms of resistance, both genetic and epigenetic1, and their existence has an evolutionary logic2.
And we really shouldn’t be surprised. Escaping from potentially lethal challenge has been a major driving force in evolution. The majority of species on Earth are parasites and their success depends on immune evasion or disguise. Potential prey similarly adopt tactics such as poisons and camouflage to thwart predators. Microbial organisms, plant seeds and some more complex animals opt for lengthy shut down or hibernation under very unfavourable circumstances. Evolution hugely favours resistance and survival.
The ‘classical’ mechanism for chemo-resistance in cancer involves the survival and then emergence of a propagating cell, or stem cell3, that has a mutation somewhere in the drug response pathway. That mutation can be in the drug’s principal target or substrate, or downstream in apoptosis effectors (e.g.p53). The principle involved is essentially the same as that first revealed for antibiotic resistance by Luria and Delbrüch in their famous fluctuation experiment with bacterial cultures. They demonstrated, by a mathematical argument, that resistance mutations pre-dated exposure to the drug or antibiotic. In other words, the drug selected for mutants rather than induced them.
The evolutionary principle could not be simpler – it’s survival of the fittest and the luckiest (Figure 1). A striking validation of this clonal selection explanation for antibiotic resistance was provided a few years ago, when it was found that some bacteria that had been frozen in permafrost for 30,000 years had mutations giving them resistance to modern antibiotics4. Similarly, members of the isolated Amazonian Yanomami tribe carry functional resistance mutations to modern, synthetic antibiotics5.
Figure 1.
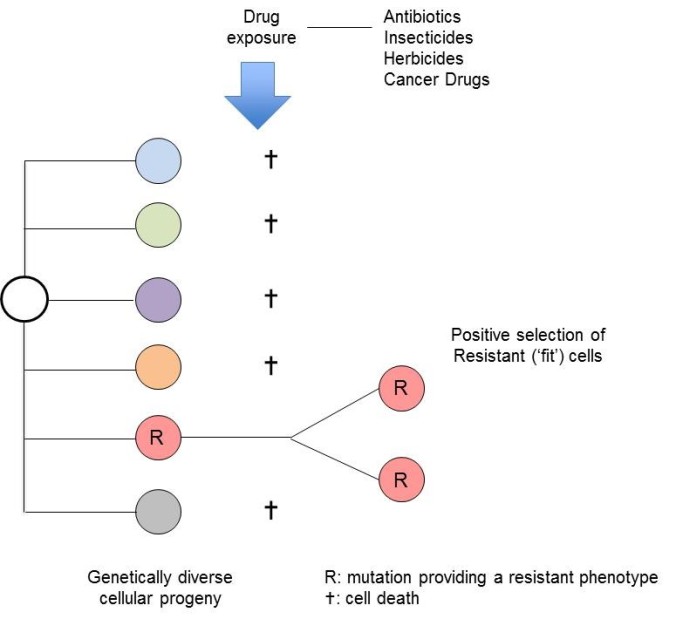
Antibiotic-resistant bacteria are now a worldwide medical problem – the blame being pinned on excessive use clinically and in animal farming, both of which provide positive selective pressure for the emergence of clones with escape or resistance mutations. Not surprisingly, the same process of resistance selection applies to insecticides and herbicides. Similarly, influenza and other viruses escape the predation of our immune systems via mutational variation.
That this problem should be so prevalent in cancer, tuberculosis, malaria or HIV/AIDS6 is the inevitable consequence of a numbers game.
Given that cells and micro-organisms both divide and mutate, the likelihood of the existence of a mutation endowing resistance is going to be determined by the clone size (number of cells) and the mutation rate. This is why small or ‘early’ tumours are less likely to appear to be resistant. When bacteria are subjected to high levels of stress, they adopt a mutator phenotype which greatly increases the probability of mutations favouring survival7. By increasing background mutation rate, the instigation of genetic instability in cancer increases the chance of drug-resistant mutations.
The prediction that escape mutations pre-date exposure to cancer drugs and that drug treatment would, essentially, positively select emergent clones with these mutations has now been validated with high depth sequencing of diagnostic samples from patients who later relapsed on targeted drug therapy8,9.
It is perhaps one of the ironies of cancer chemotherapy that intensive drug treatment provides strong selective pressure for the emergence of more robust clones10.
But the classic escape trick is not the only one available to cancer cells (Table 1). Many cancer drugs kill cancer cells by causing DNA damage and by stimulating apoptosis. But that same cell death mechanism is frequently corrupted by mutations as part of the evolutionary adaptive process of clonal evolution in cancer. Examples would be the very common loss of function deletions or mutations in p53 in cancer and the activation of BCL2 in follicular lymphoma. These mutant genotypes provide intrinsic resistance to genotoxic treatments that compromise cancer cells via cell cycle arrest and/or apoptosis.
Table 1. Evolutionary origins of selection for drug resistance in cancer
| Mechanism |
Origins |
| A. Genetics |
| • Selection of cells with pre-existing, stochastic mutations in drug response pathway |
The ‘classical’ escape route: the inevitable consequence of mutation rates and clone size. |
| • Intrinsic blocks to DNA repair / cell apoptosis pathways used by drugs |
Prior selection of acquired mutations as adaptive for cell transformation or clonal progression. |
| • Signal block bypass |
Selection of sub-clones with mutations that bypass blocked signalling with targeted therapy. |
| • Segregation of drug targets in sub-clones |
For targeted therapy. The consequence of genetic, sub-clonal variegation. |
| B. Epigenetics |
| • Quiescent stem cells |
An ancient survival trait – ‘hunkering down’ to protect against stress. Evident in bacteria and normal stem cells. |
| • Signal block bypass |
For targeted therapy. A consequence of signal network complexity, redundancy and plasticity. An early evolutionary innovation – as seen in bacteria and yeast. |
Table adapted from reference 2.
When so-called targeted therapy is used, a presumption is made that all cancer cells express the target. In some cases this may be true – for example with imatinib ABL kinase inhibitor in chronic myeloid leukaemia (CML). But more generally in advanced cancer or leukaemia, clones have a variegated genetic architecture2. A consequence is that although all cells and all clones retain the initiating, founder or truncal lesion(s), many subsequently generated mutations will be segregated into sub-clones.
Directing therapy at a mutation that is in a branch of the clonal phylogenetic tree, rather than truncal, may have an impact with shrinkage of the tumour but it is unlikely to be sustained or permanent11. Paradoxically, the demise of major drug sensitive clones will clear space and create opportunity for smaller, less competitive sub-clones that are then positively selected.
Phenotypic, or epigenetic plasticity of cancer cells provides another route to resistance 12,13. This escape ploy also exploits evolutionarily ancient survival tactics.
When bacteria, unicellular organisms and some more complex animals are confronted with harsh conditions, one survival tactic is to ‘hunker-down’ and enter a dormant or quiescent state – to re-emerge later when the coast is clear. And that may be many years, or even decades, later14.
Normal stem cells are both vital to life and limited in number. They maintain their integrity by residing predominantly in a dormant or Go cycle state15. Almost all proliferative expansion of cells (1011 per day in the marrow and small intestine) occurs in downstream progenitor or precursor cells. Stem cells enter into active cycling upon regenerative demand, which is usually transient.
Cancer stem cells can and do adopt a quiescent lifestyle. In this condition, they are largely drug and radiation resistant16. The mechanism is not entirely clear but may involve protection via physical association with stromal cells in protective niches and/or over-expression of drug efflux pumps. Presumably they are exploiting the ancient survival strategy of normal stem cells and our unicellular ancestors.
Another epigenetic mechanism of escape is particularly evident with some targeted therapies, e.g. EGFR and B-RAF inhibitors, and androgen blockade17. This again has an evolutionary logic. Cancer cells have a highly complex intracellular signalling network, albeit corrupted from normal. Similarly complex metabolic or signalling networks were an early evolutionary innovation in bacteria and are well documented in yeast cells (as shown in Figure 2, below)18.
Nodes in the pathway (Figure 2) are critical regulatory sites in the network and mutations encoding proteins in the nodes are frequently selected in cancer. But (not surprisingly) evolution has built in redundancy, fail-safe and feedback loops that can compensate for, or bypass, blocked or compromised key proteins in the network. So, with some targeted therapies, cells select for compensatory signalling which then effectively neutralises that drug-induced signalling block. This may even include a dramatic identity switch in the lineage or differentiation phenotype of the cancer cell19.
Figure 2.

Adapted from the original. Copyright © 2010, American Association for the Advancement of Science.
The image, mirroring a transcontinental airline route map, is a map of some five million binary genetic interactions mapped in yeast cells18.
Cancer cells are therefore well equipped with escape routes, and will exploit any that are accessible. When individual cancers are confronted with intensive selective pressure from targeted therapy, e.g. androgen blockade in prostate cancer, EGF receptor blocker in colorectal cancer, sub-clones independently develop resistance by co-opting one or more of the above genetic and epigenetic mechanisms20,21. The one important caveat is that resistance, whatever the mechanism, has to be expressed in a cell with self-renewal or stem cell capacity for it to have a real clonal fitness advantage and clinical impact2,3.
So, what to do about the resilience and resistance of most cancer clones? I suggest a simple traffic light analogy for thinking about this problem2. This is principally to emphasise the well-recognised fact that many, and in my view most, cancers are either inherently preventable (red light) or curable if detected early enough (red plus amber light).
The major therapeutic challenge in oncology is how to tackle drug resistance in cancers that present relatively late in their evolutionary trajectory when they are up and running (green light), e.g. glioblastoma, lung, pancreas and ovarian cancer.
Here, we need to be smarter and could doubtless learn some lessons from the non-cancer medical fields – particularly research into tuberculosis, malaria and HIV/AIDS, where drug combinations often thwart resistance6,22. Single cancer drugs will almost inevitably result in resistance, which is why combinatorial therapy is now increasingly favoured23. Quiescent cancer stem cells could be teased out of their slumber with appropriate growth factors.
Another promising idea derived from microbial studies24,25 is evolutionary steering, where appropriate scheduling and dosage of drugs can push cells into a more benign evolutionary trajectory or lower fitness cul-de-sac26,27.
Resistance to cancer chemotherapy is the consequence of evolutionary resilience – a four billion year legacy. The surprise is not drug resistance but the fact that some disseminated and intrinsically lethal cancers are drug sensitive and curable – the best examples being childhood acute lymphoblastic leukaemia, testicular cancer and choriocarcinoma. But that’s another story.
Mel
References
-
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714-726.
-
Greaves M. Evolutionary Determinants of Cancer. Cancer discovery. 2015;5(8):806-820.
-
Fong CY, Gilan O, Lam EY, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525(7570):538-542.
-
D’Costa VM, King CE, Kalan L, et al. Antibiotic resistance is ancient. Nature. 2011;477(7365):457-461.
-
Clemente JC, Pehrsson EC, Blaser MJ, et al. The microbiome of uncontacted Amerindians. Sci Adv. 2015;1(3).
-
Goldberg DE, Siliciano RF, Jacobs Jr WR. Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell. 2012;148:1271-1283.
-
Lambert G, Estevez-Salmeron L, Oh S, et al. An analogy between the evolution of drug resistance in bacterial communities and malignant tissues. Nat Rev Cancer. 2011;11(5):375-382.
-
Diaz Jr LA, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537-540.
-
Pfeifer H, Wassmann B, Pavlova A, et al. Kinase domain mutations of BCR-ABL frequently precede imatinib-based therapy and give rise to relapse in patients with de novo Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood. 2007;110(2):727-734.
-
Read AF, Day T, Huijben S. The evolution of drug resistance and the curious orthodoxy of aggressive chemotherapy. Proc. Natl. Acad. Sci., USA. 2011;108 Suppl 2:10871-10877.
-
McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7(283):283ra254.
-
Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer. 2014;14(11):747-753.
-
Holzel M, Bovier A, Tuting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer. 2013;13(5):365-376.
-
Lewis K. Persister cells, dormancy and infectious disease. Nat Rev Microbiol. 2007;5(1):48-56.
-
Wilson A, Laurenti E, Oser G, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118-1129.
-
Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834-846.
-
Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505-509.
-
Costanzo M, Baryshnikova A, Bellay J, et al. The genetic landscape of a cell. Science. 2010;327(5964):425-431.
-
Lin S, Luo RT, Ptasinska A, et al. Instructive Role of MLL-Fusion Proteins Revealed by a Model of t(4;11) Pro-B Acute Lymphoblastic Leukemia. Cancer Cell. 2016;30(5):737-749.
-
Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15(12):701-711.
-
Russo M, Siravegna G, Blaszkowsky LS, et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer discovery. 2016;6(2):147-153.
-
Hughes D, Andersson DI. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet. 2015;16(8):459-471.
-
Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30(7):679-692.
-
Nichol D, Jeavons P, Fletcher AG, et al. Steering Evolution with Sequential Therapy to Prevent the Emergence of Bacterial Antibiotic Resistance. PLoS Comput Biol. 2015;11(9):e1004493.
-
Palmer AC, Kishony R. Understanding, predicting and manipulating the genotypic evolution of antibiotic resistance. Nat Rev Genet. 2013;14(4):243-248.
-
Basanta D, Gatenby RA, Anderson AR. Exploiting evolution to treat drug resistance: combination therapy and the double bind. Molecular pharmaceutics. 2012;9(4):914-921.
-
Enriquez-Navas PM, Kam Y, Das T, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra324.
comments powered by